Die US-amerikanische Food and Drug Administration (FDA) hat ihre höchste Alarmstufe zu einer Herzpumpe herausgegeben, die mit 49 Todesfällen und 129 Verletzungen in Verbindung gebracht wurde.
Die linksseitigen Impella-Pumpen werden zur vorübergehenden Unterstützung des Herzens eines Patienten bei Eingriffen mit hohem Risiko oder nach einem schweren Herzinfarkt eingesetzt.
Die Aufsichtsbehörde warnte jedoch davor, dass es bei falscher Anwendung eine Wand in der linken Herzkammer durchstoßen könnte.
Der Hersteller des Geräts, Abiomed, hat neue Anweisungen für die Pumpe herausgegeben.
In einer am 21. März auf der Website der FDA veröffentlichten Zusammenfassung wurde der Schritt als „schwerwiegendste Art von Rückruf“ eingestuft, da bei unsachgemäßer Verwendung des Geräts das Risiko schwerer Verletzungen oder des Todes besteht.
Die Behörde warnte, dass die Verwendung der betroffenen Pumpen auch schwerwiegende gesundheitsschädliche Folgen haben könne, darunter „Bluthochdruck, mangelnde Durchblutung und Tod“.
Es wurde jedoch hinzugefügt, dass es sich bei dem Rückruf um eine Korrektur und nicht um eine Produktentfernung handele und das Gerät auf dem Markt bleiben werde.
Die Mitteilung beziehe sich auf 66.390 Geräte, die über einen Zeitraum von zwei Jahren ab dem 10. Oktober 2021 in den USA vertrieben wurden, teilte die Agentur mit.
Das Gerät erhielt 2008 die FDA-Zulassung.
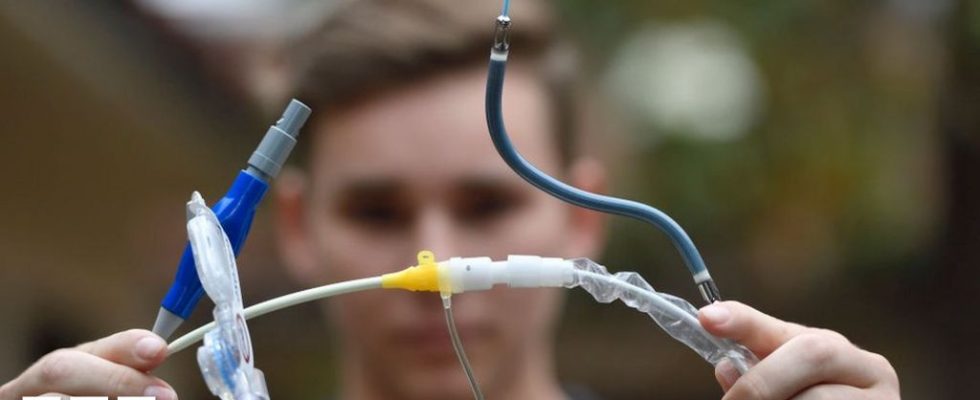
Die Pumpe verfügt über einen Katheter mit einem kleinen Haken am Ende, der durch die Blutgefäße und in die linke Herzkammer geführt wird – eine Schlüsselkammer im Herzen, die dazu dient, sauerstoffreiches Blut durch den Körper zu pumpen.
Ein Sprecher von Johnson & Johnson – das Abiomed im Jahr 2022 übernommen hat – sagte gegenüber Reuters: „Diese Mitteilung stellt keine Entfernung des Geräts dar und Impella-Herzpumpen bleiben auf dem Markt und für Patienten verfügbar.“
Abiomed habe das Risiko einer Herzperforation beim Einsetzen der Pumpen erstmals in einem technischen Bulletin im Oktober 2021 offengelegt, diese Informationen damals jedoch nicht an die FDA weitergegeben, teilte die Behörde mit.
Die Behörde führte Anfang 2023 eine Inspektion des Firmenbüros in Massachusetts durch und schickte im September ein Warnschreiben an Abiomed, in dem sie unter anderem kritisierte, dass die FDA nicht über das Risiko einer Herzperforation informiert worden sei.
Das Warnschreiben führte dazu, dass Abiomed Ende letzten Jahres ein „Urgent Medical Device Correction Letter“ herausgab, das überarbeitete Anweisungen zur korrekten Verwendung der Herzpumpe enthielt, einschließlich der Positionierung des Katheters der Pumpe oder der Verwendung von Bildgebung beim Drehen während Eingriffen, so die FDA.